research
由于RNA可以折叠成各式各样的三维结构,因而蛋白质-RNA之间形成的复合物也如蛋白质-蛋白质复合物一样复杂,蛋白质-RNA复合物结构预测与蛋白质-蛋白质复合物结构预测一样仍然很困难。因此,申请人开发了自由对接算法3dRPC (Sci Rep, 2013)与基于模板的算法PRIME(PLoS computational biology, 2016)。而基于模板的工作首次揭示了蛋白质-RNA复合物结构之间的序列-结构联系,并发现存在一个转变点。此外,通过序列分析发现了一些识别RNA结合蛋白质(RBP)的重要特征,实现了一个高精度的RBP预测算法RBPPred (Bioinformatics, 2017)。在这些工作中,我们发现RNA的结构非常重要,进一步猜测RNA结构可能在编码潜能中发挥重要作用,利用CTD编码特征来描述RNA的折叠结构,实现了RNA编码潜能评估算法CPPred(Nucleic Acids Research, 2019)。研究表明,T2,C0和GC(CTD编码的特征)在RNA编码潜能的预测上具有重要作用。CPPred在人类,小鼠,斑马鱼和酿酒酵母测试集上,具有高的准确性,然而,CPPRed在这些物种的短的RNA序列上(sORF)具有特别的优势,比之前开发的工具有一个比较大的提升。
截止2019年2月16日,申请人在PNAS、Plos computational biology、Proteins、Bioinformatics、Nucleic Acids Research等期刊上发表论文24篇,Google Scholar引用总数为667次,H-index为12。第一及通信作者论文12篇被SCI网络版摘引,被SCI引用次数为230,其中单篇被引用最高次数为62。
申请人团队目前共有博士生3名。已经毕业硕士1名和博士2名,在读博士3名。相关软件、论文等更多信息见课题组网址: http://rnabinding.com
代表成果简介:
[1] Xiaoxue Tong, Shiyong Liu*. (2019) CPPred: coding potential prediction based on the global description of RNA sequence. Nucleic Acids Research, [Epub ahead of print].
[2] Jinfang Zheng, Xiaoli Zhang, Xunyi Zhao, Xiaoxue Tong, Xu Hong, Juan Xie, and Shiyong Liu*. (2018) Deep-RBPPred: predicting RNA binding proteins in the proteome scale based on deep learning. Scientific Reports, 8(1):15264.
[3] Xiaoli Zhang; Shiyong Liu*. (2017) RBPPred: predicting RNA-binding proteins from sequence using SVM. Bioinformatics, 33(6): 854~862.
[4] Jinfang Zheng; Petras J. Kundrotas; Ilya A. Vakser*; Shiyong Liu*. (2016) Template-Based Modeling of Protein-RNA Interactions. PLoS Computational Biology, 12(9): 0~e1005120
[5] Sen Liu#, Shiyong Liu#, Xiaolei Zhu, Huanhuan Liang, Aoneng Cao, Zhijie Chang, Luhua Lai* (2007) Nonnatural protein-protein interaction-pair design by key residues grafting. PNAS, 2007 104(13):5330-35.
编码潜能预测算法
[1] Xiaoxue Tong, Shiyong Liu*. (2019) CPPred: coding potential prediction based on the global description of RNA sequence. Nucleic Acids Research, [Epub ahead of print].
成果简介:
随着人类基因组计划完成,蛋白质组学研究的开展,人们发现人类基因组中编码蛋白质的基因数目比预想中的要少很多,估计在20000~25000之间。人类基因组里面大约有3%序列编码蛋白质被转录成mRNA,其中很大部分以前认为是“垃圾”的序列也会被转录成非编码RNA(ncRNA)。寻找和指定mRNA是一项非常重要的工作。生物信息学领域由此开展了大量研究,从成千上万序列中寻觅基因(https://en.wikipedia.org/wiki/Gene_prediction),发展了比较成熟的算法。2010以前,一切都似乎尘埃落定,mRNA和非编码RNA泾渭分明。只有零星的几例研究(Rohrig et al.,PNAS,2002;Kondo et al.,Nat.Cell Biol.,2007;Galindo et al.,Plos Biology,2007)指出某些非编码 RNA 实际上是能编码的mRNA,它们包含一些较短开放阅读框(short ORFs, sORFs)。接着顶级期刊大量报道(Kondo et al.,Science,2010; Magny et al.,Science,2013;Pauli et al.,Science,2014; Anderson et al.,Cell,2015;Nelson et al.,Science,2016)长非编码RNA(lncRNA)包含编码微肽的sORF。后来发展的核糖体剖析(ribosome profiling)的新方法与 RNA 深度测序技术相结合,发现前面这些发现只是冰山一角。有大量的较短开放阅读框(sORFs)被忽视了。因此之前大部分搜寻 mRNA 的算法限于300个核苷酸以上,即至少翻译100个氨基酸。这一缺陷导致经典的基因注释软件对一些包含sORF的“长非编码RNA”进行了错误分类。RNA编码还是非编码, 这还是一个问题!
近日,我们在牛津大学出版社(Oxford University Press)出版的《核酸研究》(Nucleic Acids Research) 上发表了题为《CPPred:基于RNA序列全局描述的编码潜能预测工具》(CPPred: coding potential prediction based on the global description of RNA sequence)的研究论文,报道了一种评估RNA编码潜能的理论计算方法CPPred(Xiaoxue Tong, Shiyong Liu, Nucleic Acids Research, 2019),全文见https://doi.org/10.1093/nar/gkz087。该方法基于RNA结构可能在编码潜能中发挥重要作用这一猜测,利用CTD编码特征来描述RNA的折叠结构。研究表明,T2,C0和GC(CTD编码的特征)在RNA编码潜能的预测上具有重要作用。CPPred在人类,小鼠,斑马鱼和酿酒酵母测试集上,具有高的准确性,较目前发表的工具准确性有微弱的提高,然而,CPPRed在这些物种的短的RNA序列上(sORF)具有特别的优势,比之前开发的工具有一个比较大的提升。
利用深度学习模型对RBP进行预测
[2] Jinfang Zheng, Xiaoli Zhang, Xunyi Zhao, Xiaoxue Tong, Xu Hong, Juan Xie, and Shiyong Liu*. (2018) Deep-RBPPred: predicting RNA binding proteins in the proteome scale based on deep learning. Scientific Reports, 8(1):15264.
成果简介:
我们已经利用深度学习模型对 RBP预测进行了探索。我们把RBPPred软件包输出的物理化学特征直接输入深度学习tensorflow框架,并称之为Deep-RBPPred,相比于SVM模型,不需要计算PSSM矩阵,速度快了很多。网络结构采用的卷积神经网络。在平衡集上测试。随着训练步数的增加,预测的精度趋向于稳定,且MCC=0.74。
他人评价和国际影响:
文章发表以后,被3篇文章引用(BMC Bioinformatics201819 (Suppl 19) :516,2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM),Progress in Biochemistry and Biophysics 2018, 45(12): 1214~1223)。Zhoujian Xiao等人在文章BMC Bioinformatics201819 (Suppl 19) :516中指出深度学习在迅速发展,且在生物学领域的重要性日益增加,并提出Deep-RBPPred在蛋白质组学领域的运用。
利用SVM对RBP进行预测
[3] Xiaoli Zhang; Shiyong Liu*. (2017) RBPPred: predicting RNA-binding proteins from sequence using SVM. Bioinformatics, 33(6): 854~862.
成果简介:
通过从蛋白质的氨基酸序列出发,我们编码了一系列的物理化学属性和蛋白质的进化信息属性,具体包括:疏水性、极性、标准化的范德瓦尔斯体积、极化性、预测的二级结构、预测的溶剂可及性、侧链的带电性和极性、进化信息的位置特异性打分矩阵。在2078个RNA结合蛋白和7093个非RNA结合蛋白的训练集上采用十倍交叉验证的方式用SVM训练模型,并进行特征选择,最后在三个独立的物种测试集上进行测试。通过在不同的数据集上与其他方法进行比较,结果表明RBPPred方法的性能比目前已知的其他预测方法要好。
他人评价和国际影响:
文章发表以后,被11篇文章引用。Sarah A. Middleton等人在文章Scientific Reports volume7, Article number: 46321 (2017)中用机器学习方法预测6294个蛋白质具有RNA结合结构域,这与RBPPred预测的6657个潜在RBPs有大约2000个重合。Annkatrin Bressin等人在(http://dx.doi.org/10.1101/466151.)文章中描述RBPPred“Compared to the other earlier tools it uses comprehensive feature representation”,并且取得了和该文章种方法相似的结果。另外方法DeepMVF-RBP(BMC Bioinformatics201819 (Suppl 19) :516),利用蛋白质上物理化学性质并用深度学习方法预测RBP,结果和RBPPred相近。总的来看,目前的方法还无法完全解决RBP预测问题,还需要进一步研究。
基于自由对接的RNA-蛋白质复合物结构预测
[4] Yangyu Huang#, Shiyong Liu#, Dachuan Guo, Lin Li, Yi Xiao* (2013) A novel protocol for three-dimensional structure prediction of RNA-protein complexes Scientific reports May 28;3:1887
成果简介:
RNA-蛋白质之间的相互作用是RNA在细胞里面行使功能的重要方式之一。结构生物学家利用实验手段可以得到RNA-蛋白质复合物的三维结构, 通过原子水平的晶体结构来解释RNA与蛋白质的识别过程。 但实验取得RNA-蛋白质的复合物结构非常困难, 耗钱、耗时, 同时受限于其相互作用强度。 因而利用分子对接的方法对RNA-蛋白质复合物结构进行预测在生物医学研究中十分重要。 由于RNA可以折叠成各式各样的三维结构, 因此,RNA-蛋白质分子对接与蛋白质-蛋白质分子对接一样仍然是一个挑战。 基于快速傅里叶变换的蛋白质-蛋白质分子对接方法由以色列著名生物物理学家Katchalski-Katzir与我的博士后导师Ilya Vakser等人(Proc Natl Acad Sci U S A, 1992,2195-2199)在20年前首次提出,后经大家逐步改进,已经成为蛋白质-蛋白质分子对接领域一个主流算法。之前有人直接用蛋白质-蛋白质分子对接软件计算预测RNA-蛋白质复合物结构,成功率较低。针对这一困难,我们根据RNA-蛋白质相互作用界面存在氨基酸残基偏好、较强的静电相互作用、π-π堆积相互作用等,在我以前开发的蛋白质-蛋白质相互作用打分函数DECK(BMC Bioinformatics, 2011,280)基础上,提出了一套基于快速傅里叶变换的全新RNA-蛋白质三维复合物结构预测方法3dRPC(Huang, et al., 2013),显著地改善了预测的成功率,前10个模型的预测成功率为48%。
他人评价和国际影响:
文章发表后,在短短几个月时间内被国际同行多处正面引用 (Huang and Zou, 2014; Muppirala, et al., 2013; Tuszynska, et al., 2014)。著名生物信息学专家Janusz M. Bujnicki在综述中高度评价我们的工作:“Only recently a docking method 3dRPC was developed with a specific purpose of protein-RNA docking, which takes special features of RNA surfaces into account.”2014年,我们的方法得到了CAPRI打分竞赛知名专家Xiaoqin Zou教授等人高度评价(Huang and Zou, 2014),他们采用迭代方法得到的打分函数和我们的效果相近。总的来看,目前的方法离完全解决RNA-蛋白质复合物结构预测问题还有一定距离,特别是RNA在结合前后的构象变化问题,还需要进一步研究。
基于模板的RNA-蛋白质复合物结构预测
[5] Jinfang Zheng; Petras J. Kundrotas; Ilya A. Vakser*; Shiyong Liu*. (2016) Template-Based Modeling of Protein-RNA Interactions. PLoS Computational Biology, 12(9): 0~e1005120
成果简介:
生物物理研究所刘士勇副教授课题组最近PLOS Computational Biology上发表了在蛋白质-RNA复合物结构预测方面的研究成果,论文题目为《基于模板模建蛋白质-RNA相互作用》(Template-based modeling of protein-RNA interactions)。该工作首次揭示了蛋白质-RNA复合物结构之间的序列-结构联系,并发现存在一个转变点。同时,该研究利用这一现象开发了一套预测蛋白质-RNA复合物结构的软件PRIME(http://rnabinding.com/PRIME.html)。
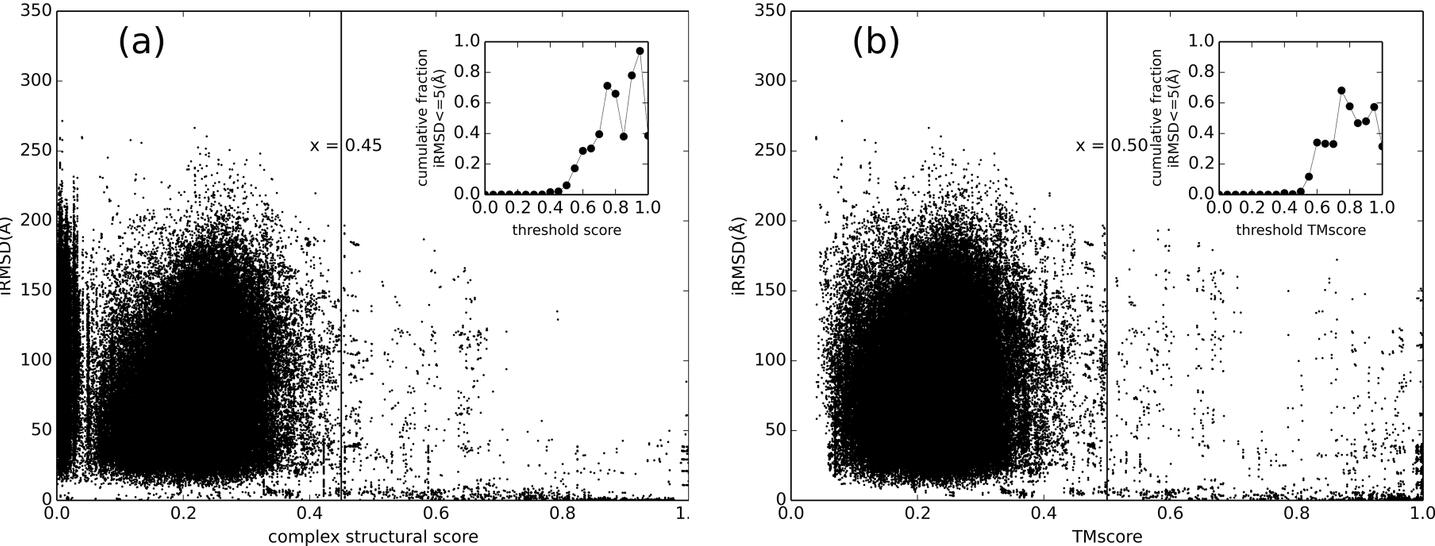
蛋白质-RNA之间的相互作用是RNA在细胞里面行使功能的重要方式之一。结构生物学家利用实验手段可以得到蛋白质-RNA复合物的三维结构, 通过原子水平的晶体结构来解释RNA与蛋白质的识别过程。但实验取得蛋白质-RNA的复合物结构非常困难, 耗钱、耗时。因而利用理论方法对蛋白质-RNA复合物结构进行预测在生物医学研究中十分重要。由于RNA可以折叠成各式各样的三维结构,因而蛋白质-RNA之间形成的复合物也如蛋白质-蛋白质复合物一样复杂,因此,蛋白质-RNA复合物结构预测与蛋白质-蛋白质复合物结构预测一样仍然很困难。而该工作提出的基于模板的方法PRIME为蛋白质-RNA复合物结构预测提供了一个全新的解决方案,经过系统比对研究发现PRIME成功率约为60%,比现有的RNA-蛋白质复合物结构预测方法要好很多的。该方法可能在基因组尺度预测非编码RNA功能、建模RNA-蛋白质相互作用方面有潜在应用价值。
物理学院博士生郑进芳为本文的第一作者,堪萨斯大学计算生物学中心Ilya Vakser教授与刘士勇副教授为共同通讯作者。此项工作得到了国家自然科学基金委、科技部863计划、中央高校基本科研业务费专项资金资助以及NIH、NSF项目的支持。
他人评价和国际影响:
文章发表以后,被5篇文章引用(Genes 2018, 9(9), 432; Methods 143 (2018) 34–47; https://doi.org/10.1101/246793; https://doi.org/10.1002/cpps.75; 10.4018/978-1-5225-2607-0.ch009),Chandran等人在文章(Genes 2018, 9(9), 432)中评论:PRIME在“自由”对接失败的情况下表现良好,并且能够解释结合时的构象变化。
针对蛋白质-蛋白质界面的功能蛋白质设计
[6] Sen Liu#, Shiyong Liu#, Xiaolei Zhu, Huanhuan Liang, Aoneng Cao, Zhijie Chang, Luhua Lai* Nonnatural protein-protein interaction-pair design by key residues grafting. PNAS, 2007 104(13):5330-35.
成果简介:
促红细胞生成素(EPO)通过和它的受体(EPOR)相互作用,促进红细胞的分化和成熟。EPO已广泛应用于临床上各种贫血的治疗。其中最有效的是肾衰、尿毒症所伴随贫血的治疗,其他对肿瘤相关性贫血,早产儿和孕产妇贫血,为手术期减少异源性输血等方面也有良好的疗效。但是用于生产重组EPO的哺乳动物细胞的培养成本很高,同时表达量不高,因而使用EPO治疗是相当昂贵的。在导师来鲁华教授的指导下,利用已建立的一套“蛋白质关键残基嫁接”算法,成功地设计了具有EPO活性的功能蛋白质,其相互作用模型细节见图1。合作者刘森等人进行的生物测活实验结果表明:设计的功能蛋白质PLC-δ1 PH结构域突变体具有拮抗EPO生物活性的功能,体外的Kd为20nM,细胞实验得到的IC50为5.7μM。
他人评价和国际影响:
该结果发表在《PNAS》上之后引起了学界关注,到目前为止,已经被引用79次,还在两本书( Ernest Giralt, Mark Peczuh, Xavier Salvatella, 2010, Protein Surface Recognition: Approaches for Drug Discovery; Andrew B. Hughes,2011,Designing New Proteins)中被引用。该PH蛋白质进行突变后与EPO只有几个功能残基一样,确具有了和EPOR结合的功能,而这种功能在自然界中是不存在的。这个例子验证了蛋白的疏水核心只起骨架的作用,功能由其局部表面决定的观点。这是首次成功地将序列上非连续的配体-受体相互作用区嫁接到非同源的骨架蛋白质上的例子。
在2011年,华盛顿大学的计算生物学学者David Baker和他的同事在《Science》上发表文章,报道了他们通过蛋白质-蛋白质分子对接方法与热点残基库(hotspot library)相结合设计出来的蛋白HB36和HB80,能够结合到广泛流行性病毒1918 H1N1蛋白血凝素(HA),并在文章中对我们的工作作为一个成功的例子进行了引用。随同发表在《Science》上的评论性的文章里,Bryan S. Der和Brian Kuhlman写道:“This strategy is reminiscent of a previous approach that involved grafting key residues from a known interaction onto a new protein scaffold togenerate a new binding pair.” 这说明David Baker小组采用的策略和我们已发表的工作(grafting key residues)比较类似。